How Do You Know Where a Gene Starts and Stops?

In computational biological science, gene prediction or factor finding refers to the process of identifying the regions of genomic Deoxyribonucleic acid that encode genes. This includes protein-coding genes as well as RNA genes, but may besides include prediction of other functional elements such every bit regulatory regions. Gene finding is 1 of the showtime and most important steps in understanding the genome of a species in one case information technology has been sequenced.
In its earliest days, "gene finding" was based on painstaking experimentation on living cells and organisms. Statistical analysis of the rates of homologous recombination of several unlike genes could determine their guild on a certain chromosome, and information from many such experiments could be combined to create a genetic map specifying the rough location of known genes relative to each other. Today, with comprehensive genome sequence and powerful computational resources at the disposal of the inquiry customs, cistron finding has been redefined every bit a largely computational trouble.
Determining that a sequence is functional should be distinguished from determining the function of the gene or its product. Predicting the function of a factor and confirming that the cistron prediction is accurate still demands in vivo experimentation[one] through gene knockout and other assays, although frontiers of bioinformatics research [ii] are making it increasingly possible to predict the office of a gene based on its sequence alone.
Gene prediction is one of the central steps in genome annotation, following sequence associates, the filtering of non-coding regions and repeat masking.[3]
Gene prediction is closely related to the and then-called 'target search trouble' investigating how Deoxyribonucleic acid-binding proteins (transcription factors) locate specific binding sites within the genome.[4] [five] Many aspects of structural cistron prediction are based on current understanding of underlying biochemical processes in the prison cell such every bit gene transcription, translation, poly peptide–protein interactions and regulation processes, which are subject area of active research in the various omics fields such as transcriptomics, proteomics, metabolomics, and more generally structural and functional genomics.
Empirical methods [edit]
In empirical (similarity, homology or bear witness-based) gene finding systems, the target genome is searched for sequences that are similar to extrinsic evidence in the form of the known expressed sequence tags, messenger RNA (mRNA), poly peptide products, and homologous or orthologous sequences. Given an mRNA sequence, it is footling to derive a unique genomic DNA sequence from which it had to accept been transcribed. Given a protein sequence, a family of possible coding DNA sequences can exist derived by opposite translation of the genetic code. In one case candidate DNA sequences take been adamant, it is a relatively straightforward algorithmic problem to efficiently search a target genome for matches, complete or fractional, and exact or inexact. Given a sequence, local alignment algorithms such as BLAST, FASTA and Smith-Waterman wait for regions of similarity between the target sequence and possible candidate matches. Matches tin can exist consummate or partial, and exact or inexact. The success of this approach is limited past the contents and accuracy of the sequence database.
A high degree of similarity to a known messenger RNA or protein product is strong show that a region of a target genome is a protein-coding gene. However, to apply this approach systemically requires extensive sequencing of mRNA and protein products. Not only is this expensive, simply in complex organisms, simply a subset of all genes in the organism'southward genome are expressed at any given time, meaning that extrinsic bear witness for many genes is not readily accessible in whatever unmarried jail cell culture. Thus, to collect extrinsic evidence for nigh or all of the genes in a complex organism requires the study of many hundreds or thousands of cell types, which presents farther difficulties. For case, some human genes may be expressed only during development as an embryo or fetus, which might exist difficult to study for ethical reasons.
Despite these difficulties, extensive transcript and protein sequence databases have been generated for human as well as other important model organisms in biological science, such as mice and yeast. For example, the RefSeq database contains transcript and protein sequence from many unlike species, and the Ensembl system comprehensively maps this testify to human and several other genomes. Information technology is, however, probable that these databases are both incomplete and incorporate small but pregnant amounts of erroneous information.
New high-throughput transcriptome sequencing technologies such as RNA-Seq and ChIP-sequencing open opportunities for incorporating additional extrinsic show into gene prediction and validation, and allow structurally rich and more accurate alternative to previous methods of measuring cistron expression such as expressed sequence tag or Deoxyribonucleic acid microarray.
Major challenges involved in gene prediction involve dealing with sequencing errors in raw DNA information, dependence on the quality of the sequence assembly, handling short reads, frameshift mutations, overlapping genes and incomplete genes.
In prokaryotes it'due south essential to consider horizontal gene transfer when searching for gene sequence homology. An additional important factor underused in current gene detection tools is being of gene clusters — operons (which are operation units of Deoxyribonucleic acid containing a cluster of genes under the control of a single promoter) in both prokaryotes and eukaryotes. Most pop gene detectors treat each cistron in isolation, independent of others, which is non biologically accurate.
Ab initio methods [edit]
Ab Initio gene prediction is an intrinsic method based on cistron content and bespeak detection. Because of the inherent expense and difficulty in obtaining extrinsic evidence for many genes, information technology is also necessary to resort to ab initio gene finding, in which the genomic DNA sequence alone is systematically searched for certain tell-tale signs of poly peptide-coding genes. These signs can exist broadly categorized as either signals, specific sequences that point the presence of a cistron nearby, or content, statistical backdrop of the protein-coding sequence itself. Ab initio cistron finding might exist more accurately characterized as cistron prediction, since extrinsic evidence is generally required to conclusively establish that a putative cistron is functional.
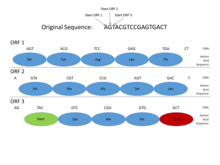
This motion picture shows how Open Reading Frames (ORFs) can be used for cistron prediction. Gene prediction is the process of determining where a coding factor might be in a genomic sequence. Functional proteins must brainstorm with a Starting time codon (where Deoxyribonucleic acid transcription begins), and end with a End codon (where transcription ends). By looking at where those codons might fall in a DNA sequence, one can see where a functional protein might be located. This is of import in gene prediction because it can reveal where coding genes are in an entire genomic sequence. In this case, a functional protein can be discovered using ORF3 because information technology begins with a Start codon, has multiple amino acids, and so ends with a Stop codon, all within the same reading frame.[6]
In the genomes of prokaryotes, genes have specific and relatively well-understood promoter sequences (signals), such as the Pribnow box and transcription factor binding sites, which are easy to systematically identify. As well, the sequence coding for a protein occurs every bit ane contiguous open reading frame (ORF), which is typically many hundred or thousands of base pairs long. The statistics of stop codons are such that fifty-fifty finding an open up reading frame of this length is a fairly informative sign. (Since 3 of the 64 possible codons in the genetic lawmaking are end codons, one would expect a stop codon approximately every 20–25 codons, or 60–75 base of operations pairs, in a random sequence.) Furthermore, protein-coding DNA has certain periodicities and other statistical properties that are easy to find in a sequence of this length. These characteristics make prokaryotic gene finding relatively straightforward, and well-designed systems are able to achieve high levels of accurateness.
Ab initio gene finding in eukaryotes, particularly complex organisms like humans, is considerably more than challenging for several reasons. First, the promoter and other regulatory signals in these genomes are more circuitous and less well-understood than in prokaryotes, making them more difficult to reliably recognize. Two classic examples of signals identified past eukaryotic factor finders are CpG islands and bounden sites for a poly(A) tail.
Second, splicing mechanisms employed past eukaryotic cells mean that a particular protein-coding sequence in the genome is divided into several parts (exons), separated by not-coding sequences (introns). (Splice sites are themselves another betoken that eukaryotic cistron finders are often designed to identify.) A typical protein-coding gene in humans might be divided into a dozen exons, each less than two hundred base pairs in length, and some as short as twenty to 30. It is therefore much more hard to detect periodicities and other known content properties of protein-coding Deoxyribonucleic acid in eukaryotes.
Advanced gene finders for both prokaryotic and eukaryotic genomes typically apply complex probabilistic models, such as subconscious Markov models (HMMs) to combine information from a diverseness of different signal and content measurements. The Blink system is a widely used and highly accurate gene finder for prokaryotes. GeneMark is another popular approach. Eukaryotic ab initio factor finders, by comparison, have achieved merely limited success; notable examples are the GENSCAN and geneid programs. The SNAP gene finder is HMM-based like Genscan, and attempts to exist more adjustable to dissimilar organisms, addressing problems related to using a gene finder on a genome sequence that information technology was not trained against.[7] A few recent approaches similar mSplicer,[viii] CONTRAST,[9] or mGene[10] also use machine learning techniques like support vector machines for successful gene prediction. They build a discriminative model using subconscious Markov support vector machines or conditional random fields to learn an authentic cistron prediction scoring office.
Ab Initio methods have been benchmarked, with some budgeted 100% sensitivity,[3] however as the sensitivity increases, accuracy suffers every bit a upshot of increased false positives.
Other signals [edit]
Amid the derived signals used for prediction are statistics resulting from the sub-sequence statistics similar grand-mer statistics, Isochore (genetics) or Compositional domain GC composition/uniformity/entropy, sequence and frame length, Intron/Exon/Donor/Acceptor/Promoter and Ribosomal binding site vocabulary, Fractal dimension, Fourier transform of a pseudo-number-coded DNA, Z-curve parameters and certain run features.[11]
Information technology has been suggested that signals other than those direct detectable in sequences may improve cistron prediction. For case, the role of secondary structure in the identification of regulatory motifs has been reported.[12] In addition, it has been suggested that RNA secondary structure prediction helps splice site prediction.[13] [fourteen] [15] [16]
Neural networks [edit]
Artificial neural networks are computational models that excel at motorcar learning and blueprint recognition. Neural networks must be trained with instance data before being able to generalise for experimental information, and tested against criterion information. Neural networks are able to come up up with approximate solutions to problems that are hard to solve algorithmically, provided there is sufficient training data. When applied to factor prediction, neural networks tin can be used alongside other ab initio methods to predict or identify biological features such equally splice sites.[17] 1 approach[eighteen] involves using a sliding window, which traverses the sequence data in an overlapping manner. The output at each position is a score based on whether the network thinks the window contains a donor splice site or an acceptor splice site. Larger windows offer more accuracy but as well require more computational power. A neural network is an example of a signal sensor equally its goal is to identify a functional site in the genome.
Combined approaches [edit]
Programs such as Maker combine extrinsic and ab initio approaches by mapping protein and EST information to the genome to validate ab initio predictions. Augustus, which may be used as part of the Maker pipeline, can as well incorporate hints in the course of EST alignments or poly peptide profiles to increment the accuracy of the gene prediction.
Comparative genomics approaches [edit]
As the unabridged genomes of many unlike species are sequenced, a promising direction in electric current enquiry on gene finding is a comparative genomics approach.
This is based on the principle that the forces of natural choice cause genes and other functional elements to undergo mutation at a slower rate than the rest of the genome, since mutations in functional elements are more probable to negatively bear on the organism than mutations elsewhere. Genes can thus exist detected by comparing the genomes of related species to find this evolutionary pressure for conservation. This arroyo was offset applied to the mouse and human genomes, using programs such equally SLAM, SGP and TWINSCAN/N-Scan and CONTRAST.[19]
Multiple informants [edit]
TWINSCAN examined but homo-mouse synteny to look for orthologous genes. Programs such equally Northward-SCAN and Contrast allowed the incorporation of alignments from multiple organisms, or in the case of N-SCAN, a single alternate organism from the target. The use of multiple informants can atomic number 82 to meaning improvements in accurateness.[19]
CONTRAST is composed of ii elements. The first is a smaller classifier, identifying donor splice sites and acceptor splice sites as well as get-go and stop codons. The second element involves constructing a full model using automobile learning. Breaking the problem into two means that smaller targeted data sets can be used to train the classifiers, and that classifier can operate independently and be trained with smaller windows. The full model can utilize the independent classifier, and non accept to waste computational fourth dimension or model complication re-classifying intron-exon boundaries. The paper in which Contrast is introduced proposes that their method (and those of TWINSCAN, etc.) be classified as de novo gene assembly, using alternate genomes, and identifying information technology equally distinct from ab initio, which uses a target 'informant' genomes.[19]
Comparative gene finding can also be used to project high quality annotations from one genome to some other. Notable examples include Projector, GeneWise, GeneMapper and GeMoMa. Such techniques now play a primal role in the note of all genomes.
Pseudogene prediction [edit]
Pseudogenes are shut relatives of genes, sharing very high sequence homology, but being unable to code for the same protein product. Whilst in one case relegated every bit byproducts of gene sequencing, increasingly, as regulatory roles are being uncovered, they are becoming predictive targets in their own right.[20] Pseudogene prediction utilises existing sequence similarity and ab initio methods, whilst adding additional filtering and methods of identifying pseudogene characteristics.
Sequence similarity methods tin can be customised for pseudogene prediction using boosted filtering to detect candidate pseudogenes. This could utilise disablement detection, which looks for nonsense or frameshift mutations that would truncate or collapse an otherwise functional coding sequence.[21] Additionally, translating DNA into proteins sequences can be more effective than only straight DNA homology.[twenty]
Content sensors can exist filtered according to the differences in statistical properties between pseudogenes and genes, such equally a reduced count of CpG islands in pseudogenes, or the differences in G-C content betwixt pseudogenes and their neighbours. Signal sensors also can be honed to pseudogenes, looking for the absence of introns or polyadenine tails. [22]
Metagenomic gene prediction [edit]
Metagenomics is the study of genetic material recovered from the environment, resulting in sequence information from a puddle of organisms. Predicting genes is useful for comparative metagenomics.
Metagenomics tools also fall into the basic categories of using either sequence similarity approaches (MEGAN4) and ab initio techniques (Glimmer-MG).
Glimmer-MG[23] is an extension to Blink that relies more often than not on an ab initio approach for gene finding and by using training sets from related organisms. The prediction strategy is augmented past classification and clustering gene information sets prior to applying ab initio gene prediction methods. The data is clustered past species. This nomenclature method leverages techniques from metagenomic phylogenetic classification. An instance of software for this purpose is, Phymm, which uses interpolated markov models—and PhymmBL, which integrates Boom into the classification routines.
MEGAN4[24] uses a sequence similarity approach, using local alignment against databases of known sequences, but also attempts to classify using additional data on functional roles, biological pathways and enzymes. Every bit in single organism cistron prediction, sequence similarity approaches are express by the size of the database.
FragGeneScan and MetaGeneAnnotator are popular gene prediction programs based on Subconscious Markov model. These predictors account for sequencing errors, partial genes and work for brusk reads.
Another fast and accurate tool for gene prediction in metagenomes is MetaGeneMark.[25] This tool is used by the DOE Joint Genome Plant to comment IMG/M, the largest metagenome collection to date.
See besides [edit]
- Listing of gene prediction software
- Phylogenetic footprinting
- Protein function prediction
- Protein structure prediction
- Protein–protein interaction prediction
- Pseudogene (database)
- Sequence mining
- Sequence similarity (homology)
References [edit]
- ^ Sleator RD (Baronial 2010). "An overview of the current status of eukaryote gene prediction strategies". Gene. 461 (1–ii): ane–4. doi:x.1016/j.gene.2010.04.008. PMID 20430068.
- ^ Ejigu, Girum Fitihamlak; Jung, Jaehee (2020-09-18). "Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing". Biological science. 9 (ix): 295. doi:10.3390/biology9090295. ISSN 2079-7737. PMC7565776. PMID 32962098.
- ^ a b Yandell Yard, Ence D (Apr 2012). "A beginner's guide to eukaryotic genome notation". Nature Reviews. Genetics. 13 (v): 329–42. doi:10.1038/nrg3174. PMID 22510764. S2CID 3352427.
- ^ Redding S, Greene EC (May 2013). "How practice proteins locate specific targets in DNA?". Chemical Physics Messages. 570: one–11. Bibcode:2013CPL...570....1R. doi:10.1016/j.cplett.2013.03.035. PMC3810971. PMID 24187380.
- ^ Sokolov IM, Metzler R, Pant K, Williams MC (August 2005). "Target search of N sliding proteins on a Deoxyribonucleic acid". Biophysical Journal. 89 (2): 895–902. Bibcode:2005BpJ....89..895S. doi:10.1529/biophysj.104.057612. PMC1366639. PMID 15908574.
- ^ Madigan MT, Martinko JM, Bender KS, Buckley DH, Stahl D (2015). Brock Biology of Microorganisms (14th ed.). Boston: Pearson. ISBN9780321897398.
- ^ Korf I (May 2004). "Gene finding in novel genomes". BMC Bioinformatics. 5: 59. doi:x.1186/1471-2105-5-59. PMC421630. PMID 15144565.
- ^ Rätsch G, Sonnenburg S, Srinivasan J, Witte H, Müller KR, Sommer RJ, Schölkopf B (February 2007). "Improving the Caenorhabditis elegans genome annotation using machine learning". PLOS Computational Biology. 3 (2): e20. Bibcode:2007PLSCB...3...20R. doi:ten.1371/journal.pcbi.0030020. PMC1808025. PMID 17319737.
- ^ Gross SS, Do CB, Sirota One thousand, Batzoglou South (2007-12-xx). "CONTRAST: a discriminative, phylogeny-free approach to multiple informant de novo gene prediction". Genome Biology. 8 (12): R269. doi:10.1186/gb-2007-8-12-r269. PMC2246271. PMID 18096039.
- ^ Schweikert G, Behr J, Zien A, Zeller G, Ong CS, Sonnenburg Southward, Rätsch G (July 2009). "mGene.web: a web service for accurate computational cistron finding". Nucleic Acids Research. 37 (Spider web Server issue): W312–six. doi:10.1093/nar/gkp479. PMC2703990. PMID 19494180.
- ^ Saeys Y, Rouzé P, Van de Peer Y (February 2007). "In search of the small ones: improved prediction of brusk exons in vertebrates, plants, fungi and protists". Bioinformatics. 23 (iv): 414–20. doi:10.1093/bioinformatics/btl639. PMID 17204465.
- ^ Hiller M, Pudimat R, Busch A, Backofen R (2006). "Using RNA secondary structures to guide sequence motif finding towards unmarried-stranded regions". Nucleic Acids Inquiry. 34 (17): e117. doi:10.1093/nar/gkl544. PMC1903381. PMID 16987907.
- ^ Patterson DJ, Yasuhara Yard, Ruzzo WL (2002). "Pre-mRNA secondary construction prediction aids splice site prediction". Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing: 223–34. PMID 11928478.
- ^ Marashi SA, Goodarzi H, Sadeghi Grand, Eslahchi C, Pezeshk H (February 2006). "Importance of RNA secondary structure information for yeast donor and acceptor splice site predictions by neural networks". Computational Biology and Chemistry. 30 (1): 50–7. doi:10.1016/j.compbiolchem.2005.10.009. PMID 16386465.
- ^ Marashi SA, Eslahchi C, Pezeshk H, Sadeghi K (June 2006). "Impact of RNA construction on the prediction of donor and acceptor splice sites". BMC Bioinformatics. 7: 297. doi:x.1186/1471-2105-seven-297. PMC1526458. PMID 16772025.
- ^ Rogic, South (2006). The office of pre-mRNA secondary construction in gene splicing in Saccharomyces cerevisiae (PDF) (PhD thesis). Academy of British Columbia. Archived from the original (PDF) on 2009-05-30. Retrieved 2007-04-01 .
- ^ Goel N, Singh S, Aseri TC (July 2013). "A comparative analysis of soft computing techniques for gene prediction". Analytical Biochemistry. 438 (1): 14–21. doi:10.1016/j.ab.2013.03.015. PMID 23529114.
- ^ Johansen, ∅Ystein; Ryen, Tom; Eftes∅l, Trygve; Kjosmoen, Thomas; Ruoff, Peter (2009). Splice Site Prediction Using Bogus Neural Networks. Computational Intelligence Methods for Bioinformatics and Biostatistics. Lec Not Comp Sci. Vol. 5488. pp. 102–113. doi:10.1007/978-3-642-02504-4_9. ISBN978-three-642-02503-7.
- ^ a b c Gross SS, Exercise CB, Sirota M, Batzoglou South (2007). "Dissimilarity: a discriminative, phylogeny-free approach to multiple informant de novo gene prediction". Genome Biology. 8 (12): R269. doi:10.1186/gb-2007-8-12-r269. PMC2246271. PMID 18096039.
- ^ a b Alexander RP, Fang G, Rozowsky J, Snyder Yard, Gerstein MB (Baronial 2010). "Annotating not-coding regions of the genome". Nature Reviews. Genetics. 11 (eight): 559–71. doi:10.1038/nrg2814. PMID 20628352. S2CID 6617359.
- ^ Svensson O, Arvestad 50, Lagergren J (May 2006). "Genome-wide survey for biologically functional pseudogenes". PLOS Computational Biological science. 2 (5): e46. Bibcode:2006PLSCB...2...46S. doi:10.1371/journal.pcbi.0020046. PMC1456316. PMID 16680195.
- ^ Zhang Z, Gerstein M (August 2004). "Large-scale analysis of pseudogenes in the human genome". Current Stance in Genetics & Development. 14 (four): 328–35. doi:10.1016/j.gde.2004.06.003. PMID 15261647.
- ^ Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL (January 2012). "Gene prediction with Glimmer for metagenomic sequences augmented past nomenclature and clustering". Nucleic Acids Research. 40 (one): e9. doi:10.1093/nar/gkr1067. PMC3245904. PMID 22102569.
- ^ Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC (September 2011). "Integrative analysis of ecology sequences using MEGAN4". Genome Enquiry. 21 (9): 1552–60. doi:x.1101/gr.120618.111. PMC3166839. PMID 21690186.
- ^ Zhu W, Lomsadze A, Borodovsky M (July 2010). "Ab initio gene identification in metagenomic sequences". Nucleic Acids Research. 38 (12): e132. doi:10.1093/nar/gkq275. PMC2896542. PMID 20403810.
External links [edit]
- Augustus
- FGENESH
- GeMoMa - Homology-based gene prediction based on amino acid and intron position conservation as well every bit RNA-Seq data
- geneid, SGP2
- Glimmer Archived 2011-08-26 at the Wayback Auto, GlimmerHMM Archived 2011-08-18 at the Wayback Machine
- GenomeThreader
- ChemGenome
- GeneMark
- Gismo
- mGene
- StarORF — A multi-platform and web tool for predicting ORFs and obtaining reverse complement sequence
- Maker - A portable and easily configurable genome annotation pipeline
Source: https://en.wikipedia.org/wiki/Gene_prediction
0 Response to "How Do You Know Where a Gene Starts and Stops?"
Enregistrer un commentaire